
Mãe busca remédio de R$ 12 milhões para salvar vida de bebê em SP: ‘Fé’
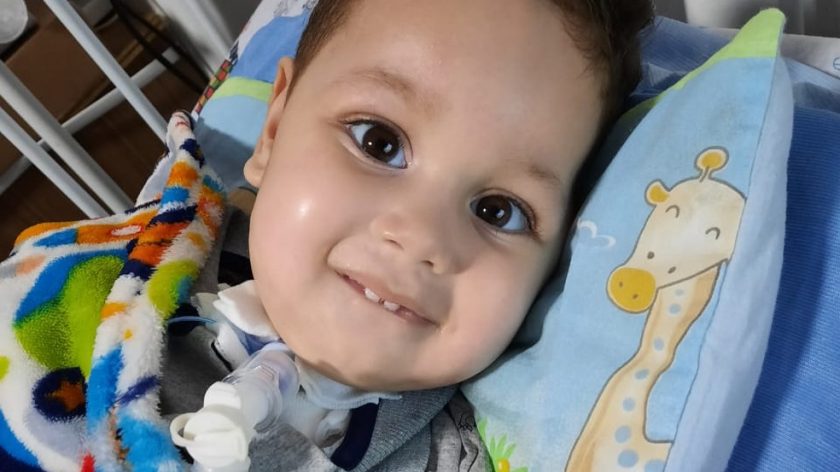

O pequeno Arthur Ferreira Belo, de apenas 1 ano e 8 meses, enfrenta uma difícil luta pela vida após ser diagnosticado com o tipo 1 da Atrofia Muscular Espinhal (AME), o mais agressivo da doença. Agora, sua família corre contra o tempo para arrecadar cerca de R$ 12 milhões e comprar o ‘remédio mais caro do mundo’, capaz de neutralizar os efeitos da doença e permitir um grande avanço no tratamento.
Para isso, foi criada uma ‘vaquinha virtual’ com o objetivo de arrecadar, até 8 de outubro, o valor necessário para importar o medicamento dos Estados Unidos. A ideia da campanha surgiu após a família de Arthur ver outros casos de vaquinhas virtuais bem sucedidas, como é o caso da Sarah e do Heitor, cujas histórias foram divulgadas pelo G1.
Segundo informações da Secretaria de Saúde do Estado, a Atrofia Muscular Espinhal é uma doença rara, degenerativa e genética, que interfere na capacidade do corpo de produzir uma proteína essencial para a sobrevivência dos neurônios motores, responsáveis pelos gestos voluntários vitais simples do corpo, como respirar, engolir e se mover. Apesar da atrofia, os pais garantem que o pequeno é muito esperto, encantador e risonho.
Arthur foi diagnosticado quando tinha apenas três meses de vida, e a notícia abalou toda a família. Isso porque a AME é a maior causa genética de morte de crianças com até 2 anos. Agora, a grande esperança dos pais é o medicamento Zolgensma, aprovado em maio de 2019 nos Estados Unidos, que promete um grande avanço na recuperação dos pacientes.
O grande problema é que esse é o remédio mais caro do mundo. Como não é vendido no Brasil, a família de Arthur precisa importá-lo dos Estados Unidos, onde chega a custar 2,125 milhões de dólares. Pela cotação atual, o medicamento pode custar R$ 11.325 milhões. Por isso, a mãe Alessandra Ferreira Santos, de 28 anos, resolveu criar uma ‘vaquinha virtual’ para arrecadar o valor.
“Temos esperança e muita fé nesse remédio. Por ser tão caro, acredito que trará um efeito positivo e que ele volte a andar. Tudo o que eu e meu marido queremos é que o nosso filho tenha a vida mais ‘normal’ possível. Mas, temos apenas três meses para arrecadar o valor, porque a medicação faz efeito somente até os dois anos, e a burocracia ainda demora. Temos muito medo de perder o Arthur”.
A família busca fazer a campanha da forma mais transparente possível. Pelas redes sociais, são postados todos os valores arrecadados por mês e quanto falta para alcançar a meta. Além disso, Alessandra e os voluntários postam outras formas de arrecadação de verba, como rifas e bingos.
Quando a campanha foi lançada, em outubro do ano passado, o dólar estava mais baixo e a família precisava de R$ 9 milhões. Agora, necessita de quase R$ 12 milhões, além da quantia necessária para pagar os custos hospitalares. “Sempre fazemos prestação de contas. Muitas pessoas acabam questionando a gente pela mudança de valor”.
Disgnóstico
Alessandra conta que, quando Arthur nasceu, percebeu que o bebê era mais ‘mole’ do que o comum, mas foi tranquilizada pelos pediatras. Mas, o tempo foi passando e, aos três meses de vida, a mãe percebeu que ele ainda não conseguia segurar o pescoço e engasgava quando mamava. Foi quando ela decidiu procurar um médico para entender o que estava acontecendo.
“Encontramos um rapaz no pronto-socorro que nos recomendou um hospital que tratava doenças raras. Fomos até lá, fizeram testes por três dias, e diagnosticaram ele com atrofia muscular. O médico disse que ele tinha uma doença rara, degenerativa, com expectativa de vida de apenas dois anos. Foi um choque, porque nunca tínhamos ouvido falar disso”, explica.
Para confirmar o diagnóstico, Arthur passou por um teste de DNA, que apontou que ele tinha o tipo 1 da doença, o mais severo. Pouco tempo depois, ele passou por duas cirurgias de traqueostomia e gastrostomia, que permitem a respiração mecânica e o suporte nutricional, respectivamente. Em seguida, o plano de saúde liberou o tratamento com o remédio Nusinersena (Spinraza).
O medicamento é o único registrado no Brasil para o tratamento da AME. Diversos estudos apontam sua eficácia na interrupção da evolução da atrofia para quadros mais graves, que são prevalentes na maioria dos pacientes. Arthur chegou a tomar seis doses do remédio, mas a evolução no tratamento continua muito lenta.
“Ele toma uma dose a cada quatro meses. Mas, se interromper, perde totalmente o efeito de tudo que já tomou. O Zolgensma é uma dose única, e pelo que acompanhamos de outras crianças, promete uma evolução muito boa no tratamento. Hoje, vivemos com medo, pois já vimos muitos pacientes morrerem por conta da doença”.
/i.s3.glbimg.com/v1/AUTH_59edd422c0c84a879bd37670ae4f538a/internal_photos/bs/2020/y/K/rUfT0hSai8CdKM5BILGQ/arthur-4.jpg)
Atualmente, Arthur mexe um pouco os braços e as pernas com a ajuda dos pais e fisioterapeutas, mas não consegue segurar objetos nem andar. Além disso, depende de ventilação mecânica 24 horas por dia, pois não consegue respirar sozinho. Com o ‘remédio mais caro do mundo’, a família busca dar maior qualidade de vida para o pequeno, que hoje sofre uma dura rotina.
De acordo com Alessandra, o medicamento vai direto para a corrente sanguínea e faz com que a proteína que não é produzida pelo paciente com AME passe a ser produzida, permitindo que os neurônios que sobreviveram possam se manter vivos e, assim, recuperar o paciente.
“Sabemos que ele não vai sair correndo assim que tomar, mas temos esperança que ele aprenda a andar e que possamos diminuir os aparelhos. É tudo muito triste, ele está crescendo e não pode brincar com as outras crianças. Esse remédio é a nossa grande esperança”.
Os pais também esperam que, quando Arthur finalmente tomar o medicamento, eles possam realizar um sonho que precisou ser adiado com o descobrimento da doença: conhecer a praia e o mar na Baixada Santista. “Nós havíamos feito muitos planos, íamos levá-lo para Mongaguá, mas os médicos não deixaram. Um dos meus sonhos é apresentar a praia para o meu filho”, finaliza.
Especialista
De acordo com a neurologista Andrea Anacleto, a Atrofia Muscular Espinhal proximal tipo 1 é uma doença rara, sem cura e que evolui de forma grave na infância, caracterizada por fraqueza muscular grave e progressiva. Segundo a especialista, a doença é ligeiramente mais frequente em meninos do que em meninas, e o diagnóstico é baseado na história clínica e exame e pode ser confirmado por teste genético.
Com a doença, o bebê geralmente apresenta fraqueza muscular grave, que afeta primeiro os braços e as pernas nas regiões proximais e depois passa para as extremidades, como mãos e pés, além de dificuldade de alimentação e insuficiência respiratória, conforme explica a neurologista.
Segundo Andrea, o ex-ministro da Saúde Luiz Henrique Mandetta assinou a incorporação do medicamento Nusinersena na Relação Nacional de Medicamentos Essenciais do Sistema Único de Saúde (SUS). Além dessa medicação, deve ser orientada uma abordagem multidisciplinar com o objetivo de melhorar a qualidade de vida.
Nos Estados Unidos, a FDA, agência que regulamenta a aprovação de medicamentos no país, autorizou a venda do remédio Zolgensma, sendo esta uma terapia gênica indicada para o tratamento da AME, usada para deter a progressão da doença. Atualmente, essa medicação não está disponível no Brasil, havendo a necessidade de que, quando indicada, seja realizada a importação.
Fonte: G1

